
In addition to the simple basic crystal structures, vampire now supports a number of more complex structures such as rock-salt such as NiO, spinels such as magnetite (Fe3O4) and Heusler alloys. These crystals are deemed complex as they each consist of more than a single material (or element). As with the simple crystals the code creates a single crystal of the chosen type. The single crystal can then be cut into the desired shape such as a nanoparticle or thin film where each of the atoms in the system can also have localized material properties. The key to using the complex crystal structures is to properly assign different materials defined in the material field to the crystal sites.
Complex crystal structures are selected in the usual way in the input file using the keyword
create:crystal-structure = <crystal type>
where crystal type can be rocksalt, spinel, heusler, or others as noted in the manual.
As with the simple crystal structures it is important to consider realistic lattice parameters and exchange constants due to the different coordination number. The definition of the unit cell size depends on the crystal structure as detailed later for each structure. A symmetric unit cell size can be selected using the key word
create:unit-cell size = 8.39 !A
which selects a size of 8.39 Angstroms in this example. The default value for the lattice constant is 3.54 Angstroms. Asymmetric unit cells such as face centred tetragonal or body centred tetragonal can be generated using directional unit cell parameters, eg
create:unit-cell-size-x = 8.62 !Acreate:unit-cell-size-y = 8.62 !Acreate:unit-cell-size-z = 8.39 !A
The complex crystal structures define multiple materials (sites) which must then be related to specific materials in the material file. This is done
using the unit-cell-category keyword:
material[1]:unit-cell-category = <ID>where <ID> refers to an arbitrary site number in the complex crystal structure. The number of sites depends on the crystal and is
2 for rock-salt, 3 for spinel and 4 for Heusler alloys. Thus, within vampire each material represents a different site symmetry and so should be
included in the material file. These are also compatible with the general geometric capabilities of vampire, allowing you to create core-shell systems
with differently doped materials, or multilayers with a single base crystal structure etc.
 Note
Note
If your material file has less materials than defined within the crystal structure, then by default those atoms are left undefined and are not generated during the creation phase.
In most cases the complex crystal structures assume the nearest neighbour approximation and so the exchange constants must be chosen to take this into account and also reflect the underlying physical properties of the system. In the following we work through a full example for CoO where both the crystal structure and the exchange interactions are somewhat complex.
Note
Some complex crystals such as spinels also include non-magnetic elements such as Oxygen. These are included in the structure for completeness, but
are explicitly excluded when calculating exchange interactions. Therefore adding exchange constants for these materials will have no effect, and the
moments will always be paramagnetic at non-zero temperature. It is always recommended that these materials are explicitly converted to non-magnetic
and removed from the system using the key word material[1]:non-magnetic = remove to avoid computational overhead when simulating them.

The rock salt crystal structure has a eight atoms per unit cell with each atom having z = 12 nearest neighbours in a face-centred-cubic configuration. Rock salt is a common structure for simple metal oxides including MgO, NiO and CoO, where oxygen atoms are modelled as non-magnetic. The crystallographic orientation is [001] along the x, y and z directions respectively. In our example we can construct a simple model of CoO. The rock salt structure can be defined using
create:crystal-structure = rocksaltcreate:unit-cell-size = 4.260 !A
which is the literature value for the lattice constant. The rock salt crystal structure in vampire defines two sublattices which must
be linked to different materials in the material file. This is done in the material file using the the commands
material[1]:unit-cell-category = 1material[2]:unit-cell-category = 2
which links material 1 to the first sublattice (unit cell category 1) and material 2 to the second sublattice (unit cell category 2). If only a single material is used then only the atoms from the first sublattice are generated. In the case of the rock salt structre each sublattice forms a face-centred-cubic lattice, and so one could conceivably model the magnetic CoO structure with an fcc crystal structure. The next step is therefore to take into account the oxygen sublattice. To do this we want to remove the atoms from the simulation, but retain them in the structure. This is done by declaring the material as non-magnetic using the key word
material[2]:non-magnetic = remove
The exchange interactions in magnetic oxides are caused by super-exchange, essentially the coupling of local magnetic moments is via polarisation of the oxygen p-orbitals. This means that the strength of the exchange interactions depends on the angle of Co-O-Co triple bonds, with the strongest antiferromagnetic bonding being for 180℃ bonds and smaller angles giving weaker antiferromagnetic exchange, and bond angles less than 90℃ being weakly antiferromagnetic.

The spinel structure is a body centred cubic structure has two atoms per unit cell and coordination number of z = 8. The crystallographic orientation is [001] along the x, y and z directions respectively. The unit cell size is defined along the cube edge and for a typical inter atomic radius of 125 pm this is 2.887 Angstroms. ε for the body centred cubic structure is 0.766. The body centred cubic structure can be defined using
create:crystal-structure = spinelcreate:unit-cell-size = 8.391 !A
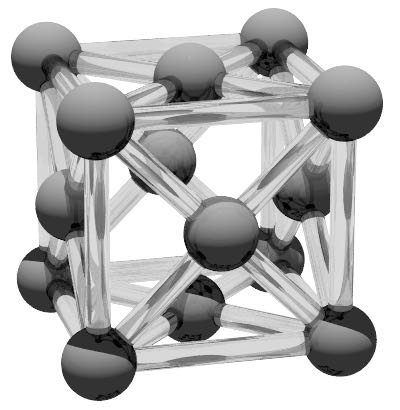
The face centred cubic structure has four atoms per unit cell and coordination number of z = 12. The crystallographic orientation is [001] along the x, y and z directions respectively. The unit cell size is defined along the cube edge and for a typical inter atomic radius of 125 pm this is 3.54 Angstroms. ε for the face centred cubic structure is 0.790. The face centred cubic structure can be defined using
create:crystal-structure = fcccreate:unit-cell-size = 3.54 !A
The hexagonal close packed structure has four atoms per unit cell and coordination number of z = 12. The crystallographic orientation is [111] along the z direction. The unit cell size is defined along the triangular edge and for a typical inter atomic radius of 125 pm this is 4.335 Angstroms. ε for the hexagonal close packed structure is 0.790. The hexagonal close packed structure can be defined using
create:crystal-structure = hcpcreate:unit-cell-size = 4.335 !A
Copyright R F L Evans 2013-2018