Vampire has a number of built in crystal structures which can be selected for your simulation. The code creates a single crystal of the chosen type to ensure that the material parameters are consistent. The single crystal can then be cut into the desired shape such as a nano particle or thin film where each of the atoms in the system can also have localized material properties.
The crystal structure is selected in the input file using the keyword
create:crystal-structure = <crystal type>
Where crystal type can be sc (simple cubic), bcc (body centred cubic), fcc (face centred cubic), or hcp (hexagonal close packed). The default is to generate a simple cubic crystal.
When choosing a crystal structure it is important to consider realistic lattice parameters and exchange constants due to the different coordination number. The definition of the unit cell size depends on the crystal structure as detailed later for each structure. A symmetric unit cell size can be selected using the key word
create:unit-cell size = 3.54 !A
which selects a size of 3.54 Angstroms in this example. The default value for the lattice constant is 3.54 Angstroms. Asymmetric unit cells such as face centred tetragonal or body centred tetragonal can be generating using directional unit cell parameters, eg
create:unit-cell-size-x = 3.62 !Acreate:unit-cell-size-y = 3.62 !Acreate:unit-cell-size-z = 3.42 !A
Different lattice constants have different coordination numbers in the nearest neighbour approximation, and so the exchange constants must be chosen to take this into account. In the nearest neighbour approximation the exchange energy Jij is given by[1]
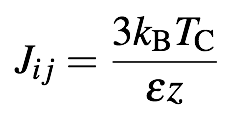
where z is the coordination number (number of nearest neighbours), Tc is the Curie temperature, kB is the Boltzmann constant, and ε is a coordination dependent spin wave stiffness factor[2], given below for different crystal structures.

The simple cubic crystal structure has a single atom per unit cell and coordination number of z = 6. The crystallographic orientation is [001] along the x, y and z directions respectively. Although no materials have a simple cubic structure, this structure is often used for atomistic 'toy models' where one wants the Heisenberg form of exchange in order to model generic temperature effects and properties. The crystal is symmetric with a lattice constant along the cube edge. ε for the simple cubic structure is 0.719. The simple cubic structure can be defined using
create:crystal-structure = sccreate:unit-cell-size = 2.40 !A
for a typical atomic radius of 125 pm.
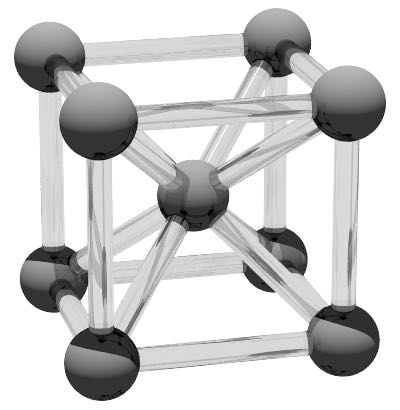
The body centred cubic structure has two atoms per unit cell and coordination number of z = 8. The crystallographic orientation is [001] along the x, y and z directions respectively. The unit cell size is defined along the cube edge and for a typical inter atomic radius of 125 pm this is 2.887 Angstroms. ε for the body centred cubic structure is 0.766. The body centred cubic structure can be defined using
create:crystal-structure = bcccreate:unit-cell-size = 2.887 !A
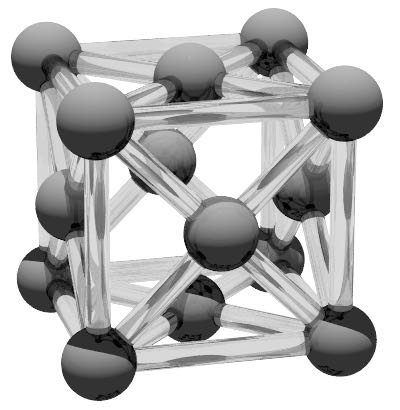
The face centred cubic structure has four atoms per unit cell and coordination number of z = 12. The crystallographic orientation is [001] along the x, y and z directions respectively. The unit cell size is defined along the cube edge and for a typical inter atomic radius of 125 pm this is 3.54 Angstroms. ε for the face centred cubic structure is 0.790. The face centred cubic structure can be defined using
create:crystal-structure = fcccreate:unit-cell-size = 3.54 !A
The hexagonal close packed structure has four atoms per unit cell and coordination number of z = 12. The crystallographic orientation is [111] along the z direction. The unit cell size is defined along the triangular edge and for a typical inter atomic radius of 125 pm this is 4.335 Angstroms. ε for the hexagonal close packed structure is 0.790. The hexagonal close packed structure can be defined using
create:crystal-structure = hcpcreate:unit-cell-size = 4.335 !A
 Note
Note
Unlike cubic crystals, the hexagonal close packed unit cell is internally assymetric, and so defining a single unit cell size will automatically generate a perfect hexagonal lattice. If different unit cell sizes are used these are related to the uniform unit cell size. For example, for a 10% compression of the crystal along the z direction one would define
create:unit-cell-size-x = 4.335 !Acreate:unit-cell-size-y = 4.335 !Acreate:unit-cell-size-z = 3.902 !A
Copyright R F L Evans 2013-2018